
Wanessa Dayanne dos Santos
Estudante de Pós Graduação da UNIFESP
Single Cell: Análise de célula única - Prática R
Explorando na Prática a Análise Transcriptoma de Células Únicas
Para explorar análises scRNA-seq aqui no Bioscritística, traremos um exemplo prático dessa abordagem através de um tutorial que abrange a análise de 2.700 células mononucleares de sangue periférico (PBMC) disponibilizado pela plataforma 10X Genomics e executado utilizando uma ferramenta bioinformática bastante comum na área, o Seurat. Antes de nos aprofundarmos no Seurat e introduzirmos os scripts das análises, é importante entendermos as etapas que envolvem a análise bioinformática dos dados scRNA-seq (Figura 5).
Essas etapas têm como objetivo obter o dado mais aproximado possível do sistema biológico estudado, e assim, minimizar os erros e interferências metodológicas.
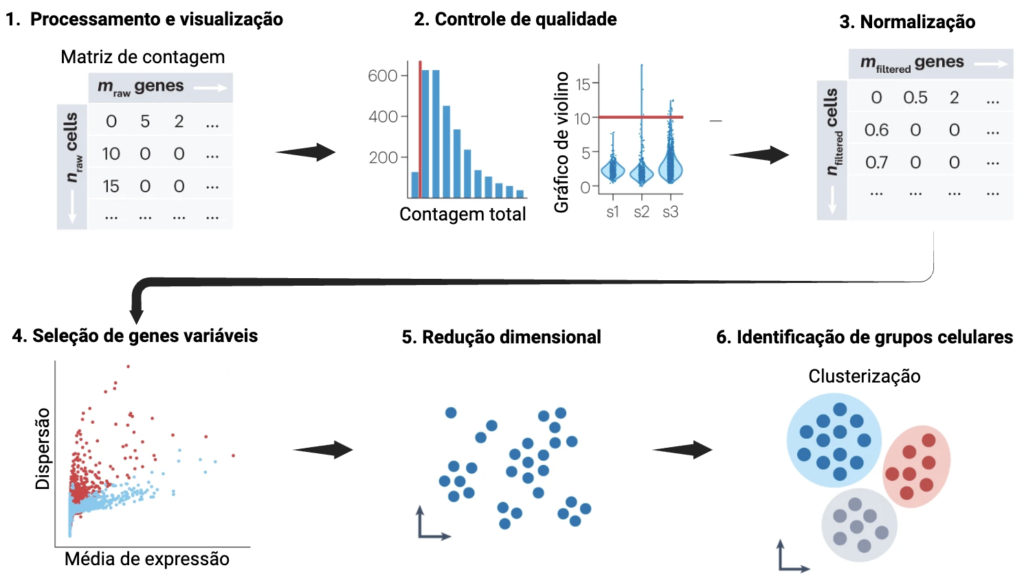
Existem diversas ferramentas de bioinformática disponíveis para execução dessas etapas. Considerando aquelas mais utilizadas nas análises de scRNA-seq, destaca-se o Seurat. O Seurat é um pacote de software de código aberto para R, desenvolvido por Satija et al. (2015), criado especificamente para a análise de dados de células individuais gerados por tecnologias de sequenciamento de nova geração. Ele engloba várias etapas do processo analítico, desde a identificação de subpopulações celulares com algoritmos de agrupamento até a descoberta de marcadores celulares. Além disso, permite a visualização de dados de alta dimensão, facilitando a interpretação dos resultados.
É importante ressaltar que o Seurat não apenas disponibiliza seu próprio conjunto de scripts, mas também permite personalizações flexíveis e a integração de ferramentas adicionais, possibilitando aprimoramentos e adaptações em cada etapa da análise conforme necessário.
Antes de começarmos a análise, é preciso garantir que o R Studio esteja instalado em seu sistema. Acesse os dados a serem utilizados aqui. Para instalar o Seurat, utilize os seguintes comandos no R (ou R Studio):
# Instalação do Seurat a partir do CRAN
install.packages('Seurat')
# Carregamento da biblioteca Seurat
library(Seurat)
# Instalação de pacotes adicionais recomendados
setRepositories(ind = 1:3, addURLs = c('https://satijalab.r-universe.dev', 'https://bnprks.r-universe.dev/'))
install.packages(c("BPCells", "presto", "glmGamPoi"))
# Instalação de pacotes adicionais usados nos tutoriais e que aprimoram a funcionalidade do Seurat
install.packages("tidyverse")
install.packages("ggplot2")
install.packages("devtools")
install.packages(c('Signac', 'SeuratData'))
remotes::install_github("satijalab/seurat-data", quiet = TRUE)
remotes::install_github("satijalab/azimuth", quiet = TRUE)
remotes::install_github("satijalab/seurat-wrappers", quiet = TRUE)
devtools::install_github("thomasp85/patchwork")
# Resolução de erros relacionados ao pacote Matrix
BiocManager::install("TFBSTools", type = "source", force = TRUE)
# Carrege as bibliotecas adicionais
library(patchwork)
library(dplyr)
library(ggplot2)
library(SeuratData)
Após a instalação bem-sucedida do Seurat e dos pacotes adicionais, podemos começar a explorar o conjunto de dados de células PBMC da 10X Genomics.
Primeiro, precisamos carregar os dados no ambiente R. Isso pode ser feito usando a função Read10X do Seurat, que lê os arquivos de saída do Cell Ranger (software da 10X Genomics) e retorna uma lista de matrizes esparsas, onde cada matriz corresponde a uma característica (em nosso caso, transcritos de RNA).
# Carregando os dados
pbmc_cells.data <- Read10X(data.dir = "/Users/Downloads/filtered_gene_bc_matrices/hg19/")
Em seguida, criamos um objeto Seurat:
pbmc_cells <- CreateSeuratObject(counts = pbmc_cells.data, project = "pbmc3k", min.cells = 3, min.features = 200)
O objeto Seurat serve como um recipiente que contém todas as informações necessárias para a análise, incluindo dados brutos, metadados e resultados de análises. Ele facilita a organização e a execução de diversas etapas da análise de células únicas de maneira eficiente.
Após a criação do objeto, o próximo passo é o Controle de Qualidade (QC). O QC é uma etapa fundamental que envolve a avaliação da qualidade das células e a remoção de possíveis outliers que podem afetar a integridade da análise. Um outlier é um valor incomum que se diferencia do padrão esperado, podendo distorcer os resultados de análises e algoritmos. Nessa fase, é comum avaliar métricas como o número de genes únicos detectados em cada célula, a contagem total de moléculas detectadas e a porcentagem de leituras que mapeiam para o genoma mitocondrial. É importante destacar que o Seurat fornece flexibilidade para explorar métricas de QC e filtrar células com base em critérios definidos pelo usuário.
No nosso caso, optamos por uma abordagem recomendada pelas melhores práticas em análise de células únicas. Utilizamos uma função personalizada chamada `mad_outlier`, baseada no Desvio Médio Absoluto (MAD), para identificar e remover outliers nos dados brutos. Essa abordagem oferece uma maneira robusta de lidar com valores discrepantes, garantindo a confiabilidade dos dados antes de prosseguirmos para etapas subsequentes da análise.
# Função para detecção de outliers usando MAD
mad_outlier <- function(vector, nmads = 5) {
median_value <- median(vector, na.rm = TRUE)
mad_value <- mad(vector, na.rm = TRUE)
upper_threshold <- median_value + nmads * mad_value
lower_threshold <- median_value - nmads * mad_value
outliers <- (vector < lower_threshold) | (vector > upper_threshold)
return(outliers)
}
# Aplicar a função mad_outlier e filtrar os dados do objeto
pbmc_cells[["percent.mt"]] <- PercentageFeatureSet(pbmc_cells, pattern = "^MT-")
pbmc_cells <- subset(pbmc_cells, subset = percent.mt < 10)
pbmc_cells$log1p_total_counts = log1p(pbmc_cells@meta.data$nCount_RNA)
pbmc_cells$log1p_n_genes_by_counts = log1p(pbmc_cells@meta.data$nFeature_RNA)
bool_vector <- !mad_outlier(pbmc_cells$log1p_total_counts, 5) &
!mad_outlier(pbmc_cells$log1p_n_genes_by_counts, 5)
pbmc_cells <- subset(pbmc_cells, cells = which(bool_vector))
Visualizando métricas de Controle de Qualidade (QC) com o gráfico de violino conforme representado na Figura 6.
VlnPlot(pbmc_cells, features = c("nFeature_RNA", "nCount_RNA", "percent.mt"), ncol = 3)
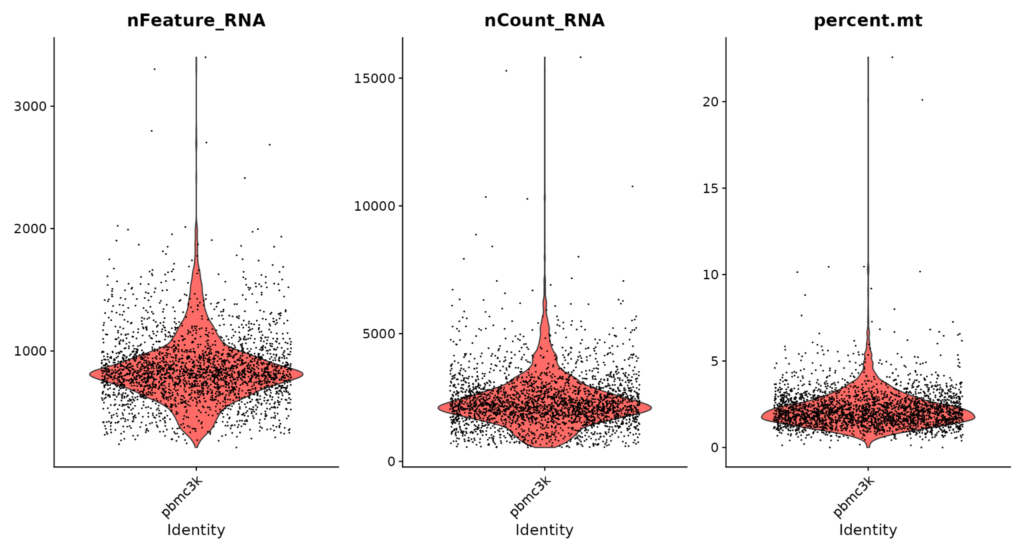
A filtragem de mitocôndrias é uma etapa importante, uma vez que as mitocôndrias são organelas celulares que têm seu próprio genoma. A presença excessiva de transcritos mitocondriais pode indicar células que estão sob estresse ou danificadas, pois essas células frequentemente apresentam aumento da atividade mitocondrial. Portanto, a filtragem de células com altos níveis de expressão de genes mitocondriais ajuda a remover essas células potencialmente problemáticas do conjunto de dados, melhorando a qualidade geral dos dados e a precisão das análises subsequentes.
Agora que realizamos a análise de qualidade das células e removemos possíveis outliers, podemos proceder com a normalização dos dados. O método padrão de normalização utilizado pelo Seurat é o “LogNormalize” , que normaliza as medições de expressão gênica para cada célula pela expressão total, multiplica esse valor por um fator de escala (10.000 por padrão) e aplica a transformação logarítmica ao resultado.
pbmc_cells <- NormalizeData(object = pbmc_cells, normalization.method = "LogNormalize", scale.factor = 10000)
A função `FindVariableGenes` é então utilizada para identificar os genes altamente variáveis, levando em consideração a média de expressão e a dispersão de cada gene. Esta etapa é fundamental para focar nos genes que realmente contribuem para a variabilidade do conjunto de dados.
pbmc_cells <- FindVariableFeatures(pbmc_cells, selection.method = "vst", nfeatures = 2000)
A seguir, é utilizado a função `ScaleData` que aplica uma transformação linear nos dados como um passo de pré-processamento antes de técnicas de redução de dimensionalidade, como a análise de componentes principais (em inglês, principal component analysis [PCA]). Ela ajusta a expressão de cada gene para que a média seja 0 e a variância seja 1 entre as células, garantindo que genes altamente expressos não dominem as análises subsequentes.
all.genes <- rownames(pbmc_cells)
pbmc_cells <- ScaleData(pbmc_cells, features = all.genes)
A análise de PCA é realizada após os dados estarem normalizados e escalados. Ela transforma um conjunto de genes que possivelmente estão correlacionados, em um conjunto menor de componentes principais não correlacionados. Para resumir, cada célula é tratada como uma individual e cada gene como uma variável. A PCA identifica os eixos nos quais a variação dos dados é máxima, esses eixos são os componentes principais.
O Seurat fornece várias maneiras de visualizar os resultados da PCA, incluindo `PrintPCA` e `PCHeatmap`.
pbmc_cells <- RunPCA(pbmc_cells, features = VariableFeatures(object = pbmc_cells))
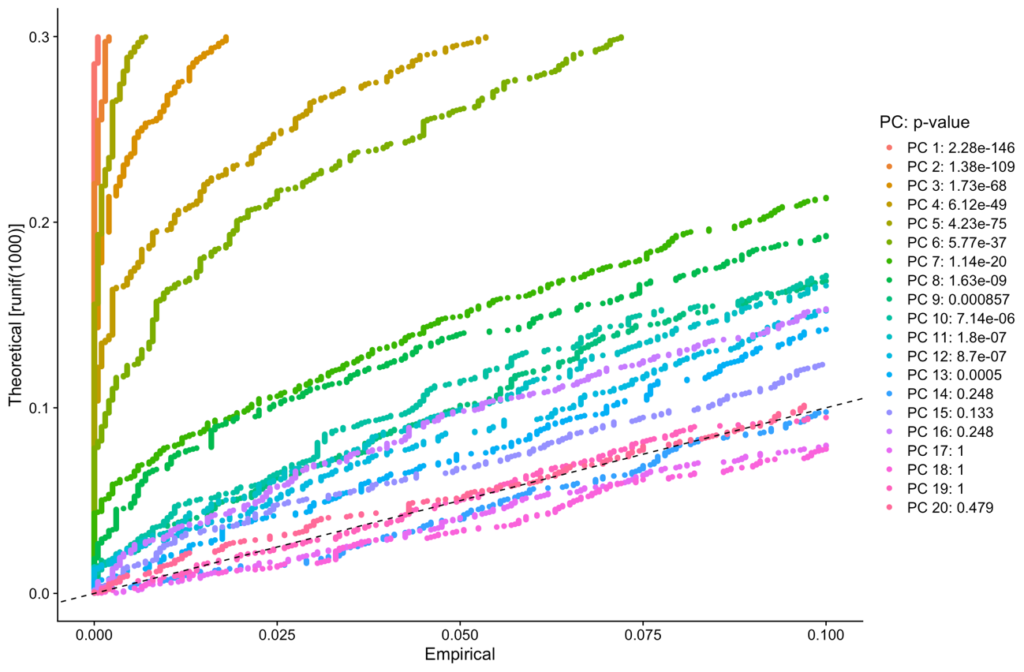
Para determinar quais PCs (do inglês, principal components) incluir nas análises posteriores, o Seurat oferece a abordagem do `JackStraw`, um teste de permutação que compara a distribuição de p-valores para cada PC com uma distribuição uniforme. A visualização do `JackStraw` ajuda a identificar PCs significativos (Figura 7).
pbmc_cells<-JackStraw(pbmc_cells,num.replicate=100)
pbmc_cells<-ScoreJackStraw(pbmc_cells,dims = 1:20)
JackStrawPlot(pbmc_cells, dims=1:20)
Outra abordagem é observar o gráfico de cotovelo (elbow plot) das variabilidades dos componentes principais. Este método costuma ser o mais utilizado devido à sua eficiência e à facilidade com que seus resultados podem ser visualizados (Figura 8).
ElbowPlot(object = pbmc_cells)
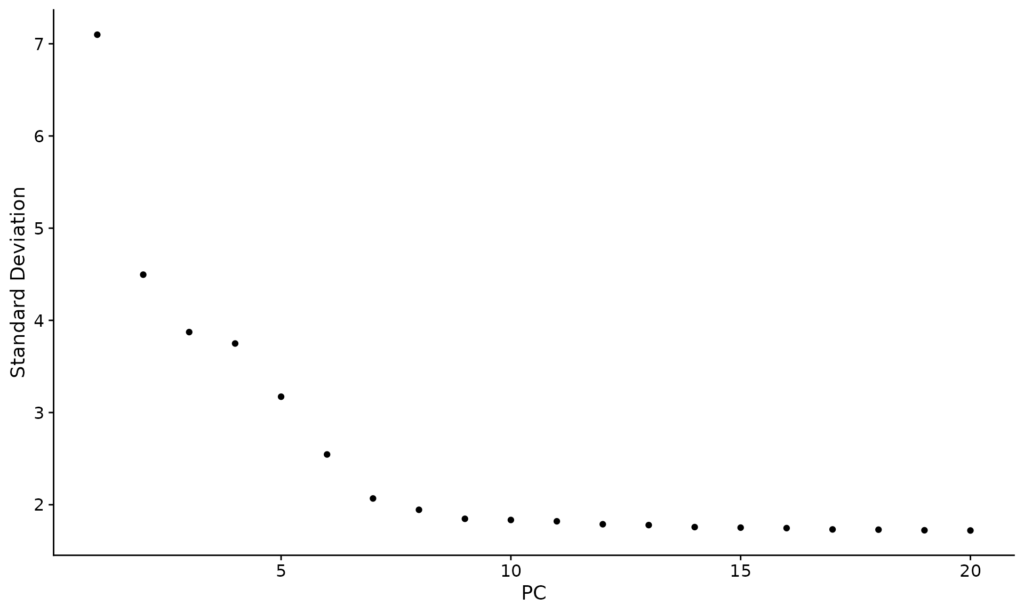
A escolha dos PCs pode então ser determinada de acordo com os valores estabelecidos nesses métodos estatísticos. No exemplo, optamos por 12 PCs com base no `Elbow plot`(Figura 8), considerando a interpretabilidade dos sinais nos PCs selecionados.
Após selecionarmos os PCs, o próximo passo é identificar grupos de células com características de transcriptomas similares. Com a função `FindNeighbors` do Seurat, é possível construir um grafo que representa as relações de proximidade entre as células no espaço do PC. Essas células são então denominadas de células vizinhas, por isso o nome da função `FindNeighbors .
pbmc_cells <- FindNeighbors(object = pbmc_cells, dims = 1:12)
Depois de identificar os vizinhos, podemos prosseguir para a etapa de clusterização, na qual agrupamos as células semelhantes com base nas informações de proximidade obtidas.
pbmc_cells <- FindClusters(object = pbmc_cells, resolution = 0.6)
O parâmetro `resolution` controla a granularidade da clusterização. Valores mais altos resultam em maiores grupos de células (em inglês clusters), enquanto valores mais baixos resultam em número menor de clusters.
Com os clusters identificados, podemos visualizar os resultados utilizando a metodologia de redução de dimensionalidade. No Seurat, a ferramenta utilizada é o UMAP (Uniform Manifold Approximation and Projection).
pbmc_cells <- RunUMAP(object = pbmc_cells, dims = 1:12)
A função `RunUMAP` calcula as coordenadas do UMAP para cada célula, incorporando informações sobre a proximidade calculada na etapa anterior. Isso nos permite visualizar a estrutura dos clusters em um espaço bidimensional. Este é um ponto essencial no processo de análise de dados de single-cell RNA-se, pois os clusters identificados podem representar diferentes tipos celulares ou estados funcionais a depender do contexto biológico do experimento.
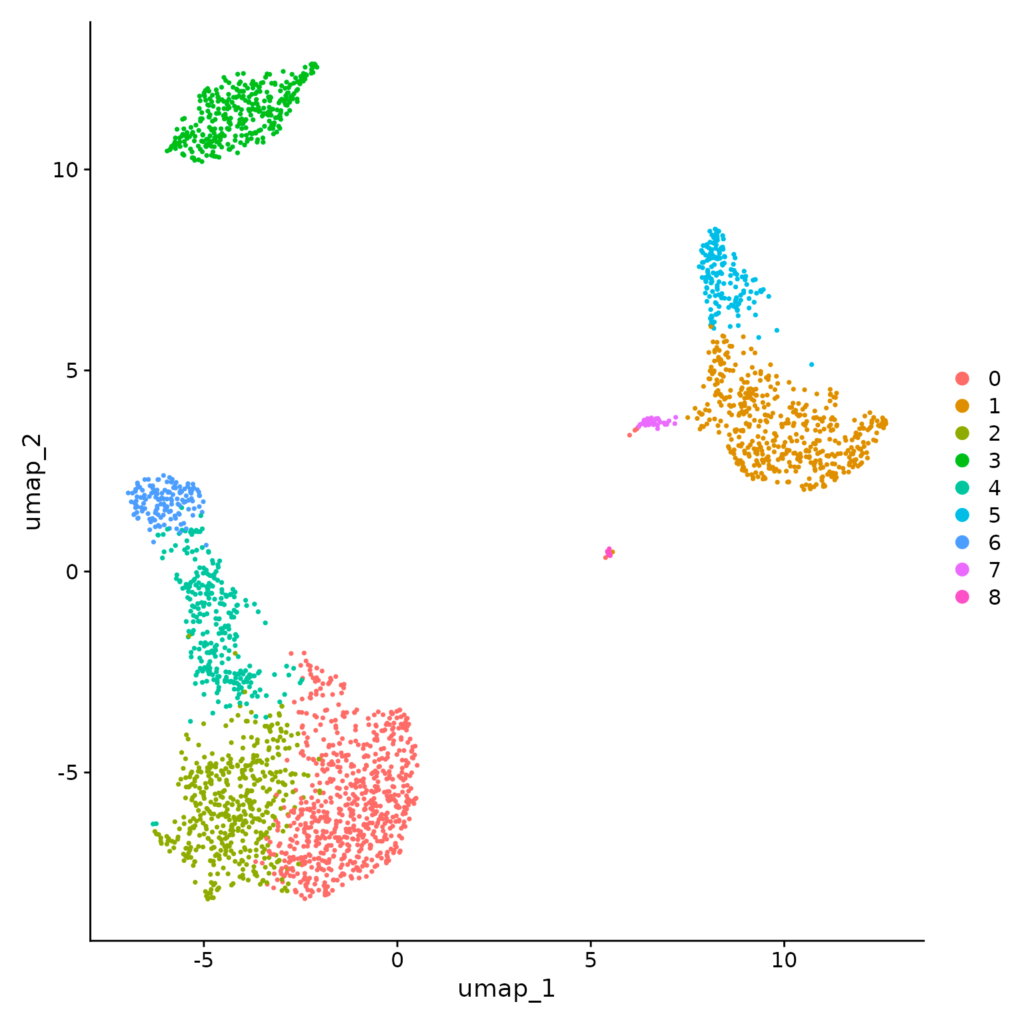
Por fim, é possível examinar e visualizar os resultados obtidos com a função `Dim Plot` (Figura 9), para representar graficamente os clusters identificados e permitindo uma interpretação mais aprofundada da heterogeneidade celular presente nos dados analisados.
DimPlot(object = pbmc_cells, group.by = "ident", cols = c("blue", "red", "green", "purple"))
A partir deste ponto, abrem-se possibilidades para muitas análises adicionais, enriquecendo a compreensão da variabilidade celular. Uma dessas análises considerada como crucial é a Análise Diferencialmente Expressa (DE), que identifica genes com uma variação significativa na expressão entre as diferentes condições experimentais. Essa abordagem é fundamental para elucidar os processos biológicos distintos em diferentes grupos celulares e a heterogeneidade presente nos dados. Vale ressaltar que, a Análise Diferencialmente Expressa (DE) não está diretamente incorporada no script principal do Seurat, uma vez que sua aplicação requer uma pergunta experimental específica. Desta forma, é preciso determinar quais elementos ou aspectos específicos que serão comparados na análise antes de prosseguir, por exemplo, identificar genes diferencialmente expressos entre diferentes tipos celulares, condições de tratamento (por exemplo, células sob diferentes estímulos ou condições ambientais) ou em resposta a perturbações específicas.
Ferramentas adicionais
Posteriormente ao processamento dos dados com o Seurat, uma série de ferramentas adicionais se tornam relevantes para as investigações seguintes. Abaixo estão algumas delas:
- CellChat:
Função Principal: Exploração de comunicação celular e interações ligante-receptor.
Criadores: Jinzhou Yuan, et al.
Data de Lançamento: 2019.
Descrição: O CellChat é uma ferramenta que destaca as interações ligante-receptor entre células únicas, proporcionando… das vias de comunicação celular presentes nos dados.
- Monocle3:
Função Principal: Inferência de trajetórias de desenvolvimento celular.
Criadores: Cole Trapnell, et al.
Data de Lançamento: Primeira versão lançada em 2020.
Descrição: O Monocle3 é especializado na inferência de trajetórias de desenvolvimento celular, permitindo a identificação de estados celulares intermediários e a análise de pseudotempo.
- SCENIC:
Função Principal: Identificação de redes regulatórias de células únicas.
Criadores: Aibar, et al.
Data de Lançamento: 2017.
Descrição: SCENIC integra expressão gênica de células únicas com dados de regiões regulatórias para identificar redes regulatórias em nível de célula única.
- Destiny:
Função Principal: Inferência de trajetórias celulares e análise de pseudotempo.
Criadores: Davide Risso, et al.
Data de Lançamento: 2018.
Descrição: Destiny é uma ferramenta para inferir trajetórias celulares e analisar pseudotempo em dados de single-cell RNA-seq.
- SingleR:
Função Principal: Comparação de perfis de expressão gênica de células únicas com referências conhecidas.
Criadores: Joshua Gould, et al.
Data de Lançamento: 2015.
Descrição: A ferramenta SingleR compara perfis de expressão gênica de células únicas com perfis de referência conhecidos, permitindo a identificação de tipos celulares e estados funcionais.
Outras técnicas de interesse em Single Cell
Exploramos a análise de transcriptoma de célula única, que nos permite investigar a expressão gênica em uma única célula. Essa técnica revolucionou a compreensão da heterogeneidade celular e da dinâmica da expressão gênica. No entanto, a expressão gênica é apenas uma de várias técnicas disponíveis quando se trata de entender a regulação gênica ao nível de célula única.
Dessa forma, podemos citar o ensaio de cromatina acessível à transposase com sequenciamento de alto rendimento (do inglês, Assay for Transposase-Accessible Chromatin using sequencing) ou ATAC-seq, uma técnica usada em biologia molecular para avaliar a acessibilidade da cromatina em todo o genoma. Ao sequenciar regiões de cromatina aberta, o ATAC-seq possibilita identificar como a condensação da cromatina e outros fatores regulatórios não codificantes influenciam a expressão gênica. Essas informações são importantes para compreender a regulação dos genes, já que a acessibilidade da cromatina pode afetar quais genes estão disponíveis para serem expressos.
Operando por meio de uma transposição controlada, este método utiliza uma enzima de transposição que integra seletivamente suas sequências adaptadoras em regiões acessíveis da cromatina. Essas regiões, presumivelmente, contêm elementos genômicos que regulam a transcrição ativa de genes, como potencializadores, promotores, locais de ligação de fatores de transcrição e entre outros (Figura 10). Após a transposição dos adaptadores, as regiões capturadas podem ser amplificadas e sequenciadas, proporcionando uma visão detalhada da acessibilidade cromatínica ao nível genômico.
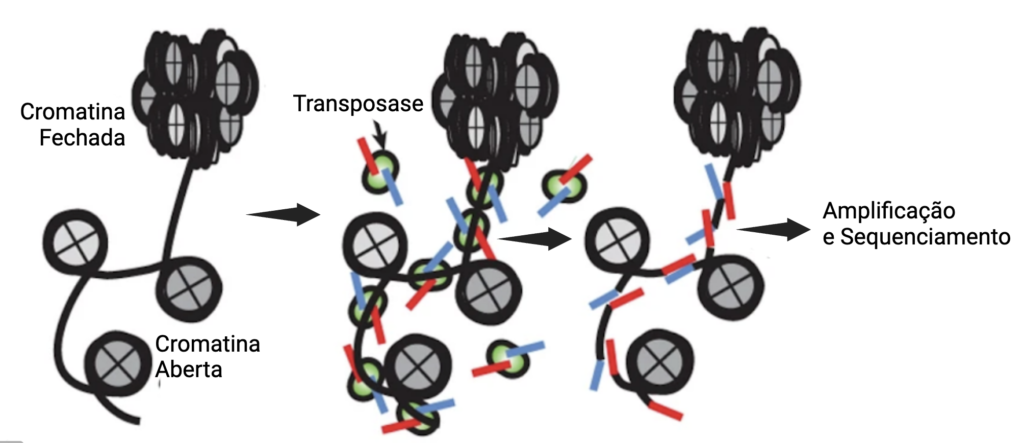
Devido à sua versatilidade, simplicidade e sensibilidade, o ATAC-seq tem superado outras técnicas semelhantes, como DNase-seq ou MNase-seq. Nos últimos anos, os protocolos de ATAC-seq foram integrados com técnicas microfluídicas, permitindo o perfilamento da cromatina aberta em resolução de célula única. Em outras palavras, ela permite aos pesquisadores ver como a estrutura da cromatina (que pode influenciar quais genes estão ativos) e a expressão gênica (quais genes estão sendo lidos e transcritos em RNA) variam de célula para célula dentro de um tecido ou organismo.
Consequentemente, o ATAC-seq tornou-se uma ótima metodologia, seja utilizada de forma independente ou em combinação com outras técnicas ômicas. O conceito de “ômica”, como já dito anteriormente, refere-se à análise global de algum aspecto biológico, como genoma (genômica), proteínas (proteômica), ou transcritos (transcriptômica). Quando falamos em abordagens multi-ômicas, estamos integrando dados provenientes de diferentes técnicas para obter uma compreensão mais completa e integrada de um sistema.
A integração do ATAC-seq com outras técnicas ômicas, como a análise de transcriptoma de célula única (scRNA-seq), possibilita uma abordagem multi-ômica para estudar a expressão gênica em nível de célula única. Enquanto o ATAC-seq revela informações sobre a acessibilidade cromatínica, o scRNA-seq fornece dados sobre a expressão gênica em células individuais. Ferramentas como o Seurat (Satija et al., 2019) também oferecem métodos para a integração dessas técnicas, permitindo correlacionar padrões de abertura cromatínica com a expressão gênica específica de cada célula, proporcionando uma visão mais completa e integrada da biologia celular em diferentes contextos experimentais.
Em conclusão, destaca-se a relevância fundamental da bioinformática. Ao aplicarmos suas ferramentas, desde o Seurat até técnicas como o ATAC-seq, percebemos a bioinformática como um pilar essencial para a interpretação de amplos conjuntos de dados. A análise integrada de informações provenientes de single-cell RNA-seq e ATAC-seq oferece novas perspectivas sobre a heterogeneidade celular, consolidando a bioinformática como uma ferramenta indispensável na era da genômica e da biologia celular.
Exercícios
a) Qual é a principal limitação da análise de RNA em massa comparada à análise de transcriptoma de célula única?
b) Explique brevemente como a metodologia baseada em microgotículas da 10x Genomics funciona no sequenciamento de RNA de células únicas e destaque uma vantagem.
c) Como a técnica MDA contribuiu para o desenvolvimento da tecnologia de Single Cell?
Referências
ANGERER, Philipp; LALEH HAGHVERDI; BÜTTNER, Maren; et al. Destiny: diffusion maps for large-scale single-cell data in R. Bioinformatics, v. 32, n. 8, p. 1241–1243, 2015. Disponível em: https://academic.oup.com/bioinformatics/article/32/8/1241/1744143. Acesso em: 20 jan. 2024.
ARAN, Dvir. SingleR. Aran Lab Technion. Disponível em: https://aran-lab.com/software/singler/. Acesso em: 11 jan. 2024.
BOLETIM DA UFMG. Ufmg.br. Disponível em: https://www.ufmg.br/boletim/bol1832/4.shtml. Acesso em: 11 jan. 2024.
CLARK, Sheila. Single cell RNA-seq: An introductory overview and tools for getting started – 10x Genomics. 10x Genomics. Disponível em: https://www.10xgenomics.com/blog/single-cell-rna-seq-an-introductory-overview-and-tools-for-getting-started. Acesso em: 11 jan. 2024.
Cole Trapnell Lab. Monocle 3. Disponível em: https://cole-trapnell-lab.github.io/monocle3/. Acesso em: 11 jan. 2024
F. ALEXANDER WOLF; ANGERER, Philipp ; THEIS, Fabian J. SCANPY: large-scale single-cell gene expression data analysis. Genome Biology, v. 19, n. 1, 2018. Disponível em: <https://genomebiology.biomedcentral.com/articles/10.1186/s13059-017-1382-0>. Acesso em: 15 jan. 2024.
HEGENBARTH, Jana-Charlotte; LEZZOCHE, Giuliana; DE, J.; et al. Perspectives on Bulk-Tissue RNA Sequencing and Single-Cell RNA Sequencing for Cardiac Transcriptomics. Frontiers in Molecular Medicine, v. 2, 2022. Disponível em: https://www.frontiersin.org/articles/10.3389/fmmed.2022.839338/full. Acesso em: 11 jan. 2024.
JIN, Suoqin; GUERRERO‐JUAREZ, Christian F; ZHANG, Lihua; et al. Inference and analysis of cell-cell communication using CellChat. Nature Communications, v. 12, n. 1, 2021. Disponível em: <https://www.nature.com/articles/s41467-021-21246-9>. Acesso em: 15 jan. 2024.
PAN, Yating; CAO, Wenjian; MU, Ying; et al. Microfluidics Facilitates the Development of Single-Cell RNA Sequencing. Biosensors, v. 12, n. 7, p. 450–450, 2022. Disponível em: https://www.mdpi.com/2079-6374/12/7/450. Acesso em: 11 dez. 2023.
PICELLI, S. (2017). Single-cell RNA-sequencing: The future of genome biology is now. RNA Biology, 14(5), 637-650.
SATIJA, R.; FARRELL, J. A.; GENNERT, D.; SCHIER, A. F.; REGEV, A. (2015). Seurat: A practical guide. Springer.
SATIJA, R.; FARRELL, J. A.; GENNERT, D.; SCHIER, A. F.; REGEV, A. (2015). Spatial reconstruction of single-cell gene expression data. Nature Biotechnology, 33(5), 495-502.
STUART, T.A; BUTLER, Andrew; HOFFMAN, Paul; et al. Comprehensive Integration of Single-Cell Data. Cell, v. 177, n. 7, p. 1888-1902.e21, 2019. Disponível em: <https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6687398/>. Acesso em: 13 jan. 2024.
Single Cell (10X Genomics) – Centre for PanorOmic Sciences (CPOS). Cpos.hku.hk. Disponível em: <https://cpos.hku.hk/portfolio-item/single-cell-10x-genomics/>. Acesso em: 8 jan. 2024.
THEMEFISHER. SCENIC. Disponível em: https://scenic.aertslab.org/. Acesso em: 11 jan. 2024.
