
Wanessa Dayanne dos Santos
Estudante de Pós Graduação da UNIFESP
Single Cell: Análise de célula única – Teoria
Bioinformática e a Análise de célula única
O campo da bioinformática emerge da interseção entre biologia e ciência da computação, desempenhando um papel central na pesquisa biológica contemporânea. Sua história remonta à década de 1950, quando o código genético do DNA começou a ser desvendado. Em 1952, experimentos conduzidos por Hershey e Chase solidificaram a compreensão do DNA como a molécula responsável pela transmissão genética em células bacterianas, e em 1953, a estrutura de dupla hélice do DNA foi elucidada por Watson, Crick e Franklin.
O impulso significativo da bioinformática surgiu nos anos 90 com o Projeto Genoma Humano que destaca a urgente necessidade de ferramentas computacionais diante da explosão de dados genômicos. Nesse cenário, a bioinformática se tornou uma disciplina obrigatória na interpretação e análise de dados biológicos, desempenhando papel fundamental na decifração dos intricados códigos genéticos.
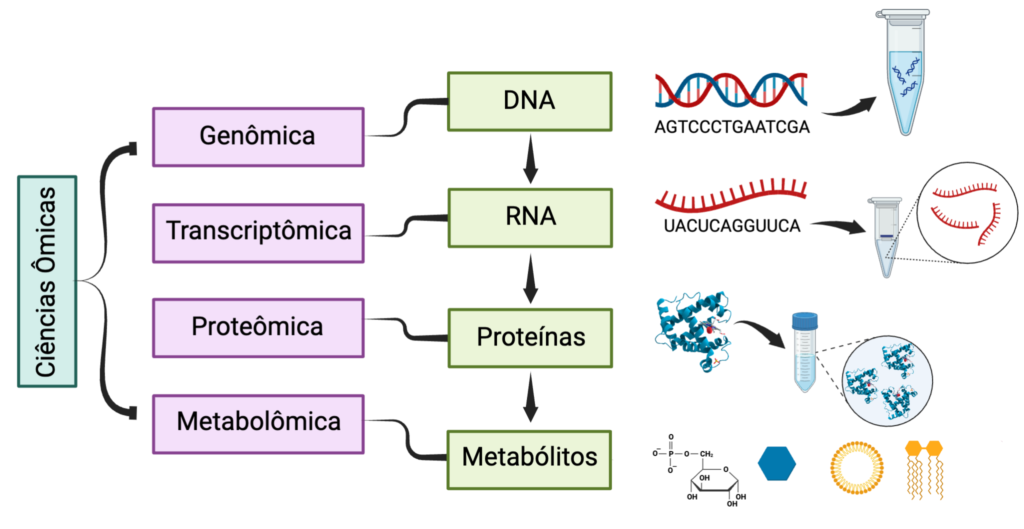
Dentro desse contexto, emergiram diversas áreas de estudo conhecidas como “ômicas”. “Omic”, derivado do latim “ome” quer dizer muitos ou em massa. Ao se referir às análises ômicas, significa que serão analisadas grandes quantidades de dados de diferentes tipos de moléculas. Cada ômica irá se concentrar em um aspecto específico da biologia molecular, temos como exemplos a genômica, que estuda o conjunto completo de genes, a transcriptômica, que analisa moléculas de RNA, a proteômica, que investiga o conjunto completo de proteínas, e a metabolômica, que analisa o conjunto de metabólitos funcionais em uma célula (Figura 1).
O aprimoramento das ferramentas experimentais e bioinformáticas, focado no sequenciamento dos transcritos de RNA (também denominado RNA-seq em inglês), foi impulsionado pelo progresso na área da transcriptômica. Esse desenvolvimento resultou em uma técnica inovadora conhecida como: análise de transcriptoma de célula única, em inglês, Single‐cell RNA sequencing (scRNA‐seq, quando abreviado). Essa técnica surgiu da necessidade de estudar o comportamento individual de cada célula em uma amostra, ultrapassando as limitações dos métodos convencionais. A análise de célula única oferece uma compreensão detalhada do transcriptoma por tipo celular, proporcionando uma compreensão mais detalhada da heterogeneidade celular.
No entanto, é preciso enfatizar que a complexidade dos dados gerados representou um grande desafio, principalmente devido à vasta quantidade de informações, demandando abordagens bioinformáticas avançadas para obter uma interpretação correta e verídica. Diante dessa dificuldade, ferramentas bioinformáticas especializadas, como Seurat (Satija et al., 2015) e Scanpy (Wolf et al., 2018), destacam-se na interpretação e análise dos dados provenientes do scRNA-seq. Essas ferramentas permitem a visualização clara dos perfis transcriptômicos, o que facilita a identificação de padrões e a diferenciação de subtipos celulares, tornando-se indispensáveis para análises desse campo.
À medida que a análise de transcriptoma de célula única amplia a escala dos dados gerados, a bioinformática responde com a criação de algoritmos e métodos analíticos específicos. Essa colaboração interdisciplinar, essencial para a interpretação desses dados complexos, impulsiona a busca por compreensões significativas no cenário biológico. Com isso, avançamos para explorar mais detalhadamente o universo da tecnologia para análise single-cell
Aprofunde o seu conhecimento
Pesquise na literatura para responder às perguntas abaixo:
a) Explique o conceito de “ômicas” mencionado no texto e como ele contribui para uma compreensão mais abrangente dos processos biológicos.
b) Como a análise de transcriptoma de célula única (scRNA-seq) supera as limitações dos métodos convencionais, conforme mencionado no texto?
c) Como a bioinformática responde ao aumento na escala dos dados gerados pela análise de transcriptoma de célula única?
Introdução à Tecnologia de Single Cell
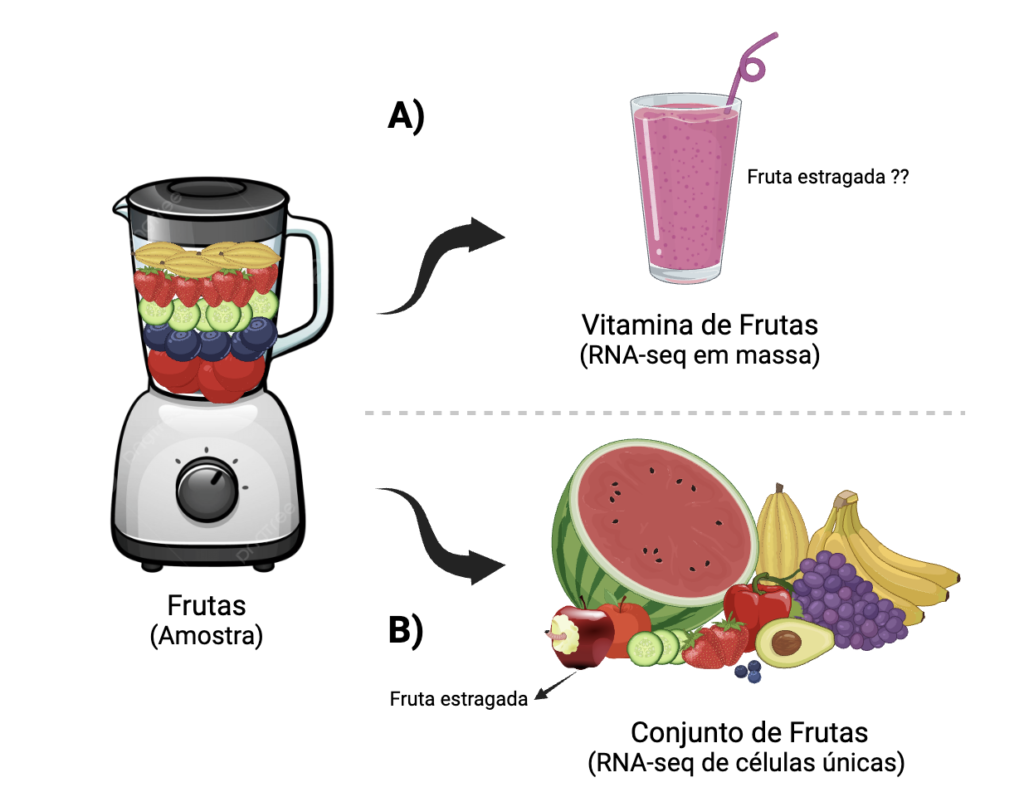
Para introduzir a tecnologia Single-Cell RNA sequencing (scRNA-seq), trazemos a analogia da vitamina de frutas (Figura 2). Imagine que há uma variedade de frutas diferentes disponíveis: maçãs, bananas, laranjas, morangos, entre outras. Se todas essas frutas forem batidas em um liquidificador e usadas para fazer uma vitamina, o processo resultará em uma mistura bem homogênea das frutas utilizadas na mistura (Figura 2A). Esse cenário pode ser comparado à análise de RNA em massa, também conhecida como “bulk RNA sequencing”. Uma metodologia que surgiu no final dos anos 2000 com o advento das técnicas de sequenciamento de próxima geração (NGS). Com essa tecnologia, não é possível sabermos exatamente quais frutas foram utilizadas para fazer uma vitamina. Deste modo, se uma das frutas estiver estragada, pode afetar o sabor geral da vitamina, e não é possível identificar exatamente qual fruta causou o problema. O desenvolvimento da tecnologia scRNA-seq vence essa limitação (Figura 2B). Com ela, é possível realizar uma análise para identificar os componentes únicos na amostra. Em nossa analogia, é possível identificar quais frutas foram utilizadas inicialmente para realizar a vitamina.
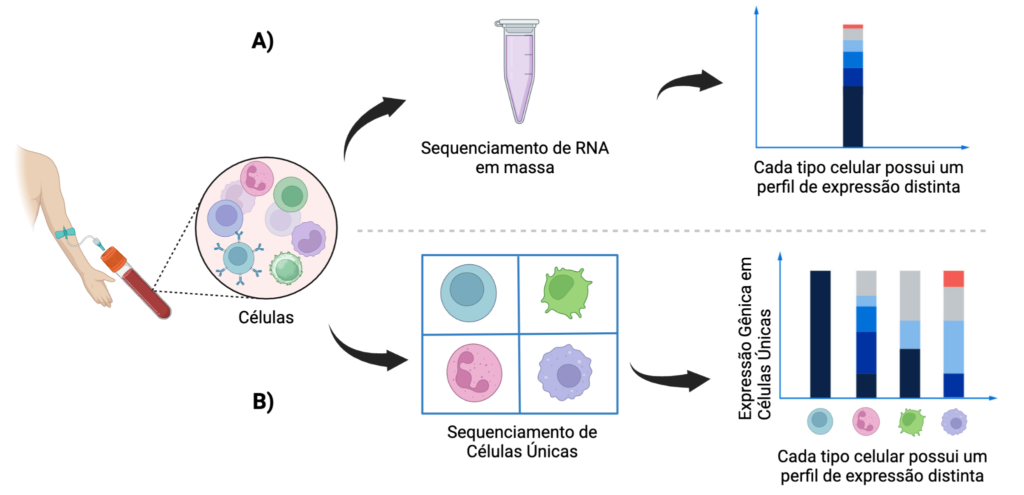
Vamos agora aprofundar no entendimento da tecnologia scRNA-seq trazendo esses conceitos para a aplicação em tecidos corporais. Pesando no tecido sanguíneo, ele é composto por diferentes tipos de células, como linfócitos T, linfócitos B, monócitos, neutrófilos, células Natural Killer (NK), células dendríticas, eosinófilos e basófilos. Utilizando RNA-seq em massa (Figura 3A), temos como resultado a expressão dos RNAs de todas as células juntas, contudo, não é possível saber o perfil expressão gênica por tipo celular. Não é possível, por exemplo, identificar os padrões de expressão gênica dos linfócitos B. Utilizando a analogia da vitamina, o RNA de todas as células é extraído e sequenciado em conjunto, resultando em uma “vitamina” de informações genéticas. Com a tecnologia scRNA-seq, é possível analisar o perfil genético de cada célula individualmente (Figura 3B).
A introdução dessa análise foi um avanço significativo que compensou as limitações da análise de RNA em massa, permitindo uma visão mais detalhada e precisa da expressão gênica em nível de célula única. Isso abriu novas possibilidades para o estudo da diversidade e complexidade celular. Voltando novamente a analogia da vitamina de frutas, se uma fruta estiver estragada e modificar o sabor da vitamina, é possível identificar qual fruta está causando esse sabor. Aplicando para os estudos biomédicos, essa tecnologia pode, por exemplo, ser utilizada para avaliar a heterogeneidade de um tumor, e ainda, possibilitar a investigação de biomarcadores para doenças complexas.
As primeiras abordagens da técnica de scRNA-seq começaram a ser desenvolvidas, marcando o início de uma revolução na análise genética em nível celular. Em 2009, a técnica denominada MDA (Multiple Displacement Amplification) foi introduzida por Dean et al., proporcionando uma maneira pioneira de amplificar o DNA de uma única célula, permitindo a análise de genomas individuais. No entanto, essas técnicas iniciais apresentavam desafios significativos, incluindo a amplificação desigual de diferentes regiões do genoma e a ocorrência de muitos erros durante o processo.
Em 2010, surgiu uma nova abordagem conhecida como MALBAC (Multiple Annealing and Looping-Based Amplification Cycles) desenvolvida por Tang et al. Essa técnica superou algumas limitações da MDA, proporcionando uma amplificação mais uniforme e reduzindo a taxa de erros. A introdução do MALBAC foi um marco na evolução das técnicas de scRNA-seq, impulsionando a aceitação e a aplicação generalizada dessas metodologias. Além disso, em 2012, Hashimshony et al. introduziram a técnica CEL-seq, oferecendo uma nova abordagem para o sequenciamento de RNA de célula única.
O sequenciamento de células únicas foi reconhecido em 2013 como o método do ano pelo Nature Publishing Group, consolidando a sua importância para a comunidade científica. Esse reconhecimento representou não apenas um avanço técnico, mas também um indicativo do crescente impacto da tecnologia de scRNA-seq na pesquisa biomédica e genômica. A evolução continuou com o surgimento de novas técnicas como a Drop-seq (Macosko et al., 2015) e a InDrops (Klein et al., 2015). Já em 2016, a plataforma Chromium 10X comercializada pela empresa 10x Genomics foi introduzida, proporcionando métodos ainda mais eficientes.
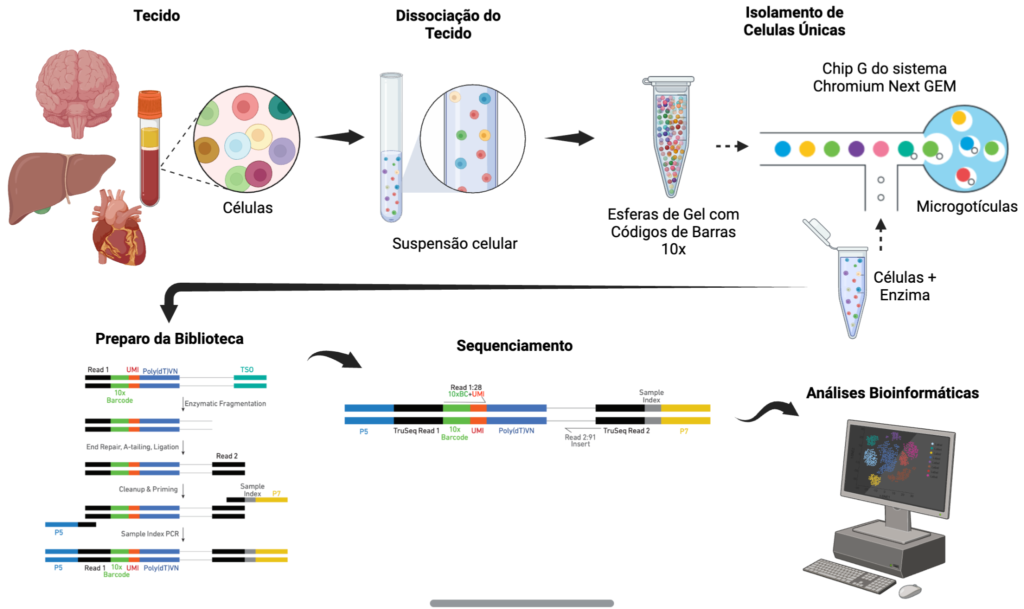
Adicionalmente, em 2019, a 10x Genomics apresentou uma metodologia baseada em microgotículas, conforme representado na Figura 4. Nesse método, cada célula individual é encapsulada em uma pequena gota contendo todos os reagentes necessários para a identificação de transcritos advindos de uma única célula. Após a célula ser rompida, ou lisada, o material genético dentro de cada gota é marcado com um código de barras único (oligonucleotídeos de sequências únicas). Esse código de barras permite que o RNA de cada célula seja amplificado e lido de forma independente. Essa tecnologia se destaca por sua eficiência na análise de expressão gênica em nível de célula única e pela capacidade de processar milhares de células simultaneamente. Atualmente, é considerada uma das metodologias mais utilizadas pelos estudos envolvendo análises de scRNA-seq.
Avançando para 2021, a técnica de análise espacial de célula única (Single Cell Spatial Transcriptomics, em inglês) surge como uma inovação notável, possibilitando a análise da expressão gênica em células individuais enquanto mantém sua localização espacial nos tecidos. Esses avanços representam apenas uma fração do progresso contínuo no campo de scRNA-seq, destacando sua relevância e potencial incessante na pesquisa biomédica.
Referências
ANGERER, Philipp; LALEH HAGHVERDI; BÜTTNER, Maren; et al. Destiny: diffusion maps for large-scale single-cell data in R. Bioinformatics, v. 32, n. 8, p. 1241–1243, 2015. Disponível em: https://academic.oup.com/bioinformatics/article/32/8/1241/1744143. Acesso em: 20 jan. 2024.
ARAN, Dvir. SingleR. Aran Lab Technion. Disponível em: https://aran-lab.com/software/singler/. Acesso em: 11 jan. 2024.
BOLETIM DA UFMG. Ufmg.br. Disponível em: https://www.ufmg.br/boletim/bol1832/4.shtml. Acesso em: 11 jan. 2024.
CLARK, Sheila. Single cell RNA-seq: An introductory overview and tools for getting started – 10x Genomics. 10x Genomics. Disponível em: https://www.10xgenomics.com/blog/single-cell-rna-seq-an-introductory-overview-and-tools-for-getting-started. Acesso em: 11 jan. 2024.
Cole Trapnell Lab. Monocle 3. Disponível em: https://cole-trapnell-lab.github.io/monocle3/. Acesso em: 11 jan. 2024
F. ALEXANDER WOLF; ANGERER, Philipp ; THEIS, Fabian J. SCANPY: large-scale single-cell gene expression data analysis. Genome Biology, v. 19, n. 1, 2018. Disponível em: <https://genomebiology.biomedcentral.com/articles/10.1186/s13059-017-1382-0>. Acesso em: 15 jan. 2024.
HEGENBARTH, Jana-Charlotte; LEZZOCHE, Giuliana; DE, J.; et al. Perspectives on Bulk-Tissue RNA Sequencing and Single-Cell RNA Sequencing for Cardiac Transcriptomics. Frontiers in Molecular Medicine, v. 2, 2022. Disponível em: https://www.frontiersin.org/articles/10.3389/fmmed.2022.839338/full. Acesso em: 11 jan. 2024.
JIN, Suoqin; GUERRERO‐JUAREZ, Christian F; ZHANG, Lihua; et al. Inference and analysis of cell-cell communication using CellChat. Nature Communications, v. 12, n. 1, 2021. Disponível em: <https://www.nature.com/articles/s41467-021-21246-9>. Acesso em: 15 jan. 2024.
PAN, Yating; CAO, Wenjian; MU, Ying; et al. Microfluidics Facilitates the Development of Single-Cell RNA Sequencing. Biosensors, v. 12, n. 7, p. 450–450, 2022. Disponível em: https://www.mdpi.com/2079-6374/12/7/450. Acesso em: 11 dez. 2023.
PICELLI, S. (2017). Single-cell RNA-sequencing: The future of genome biology is now. RNA Biology, 14(5), 637-650.
SATIJA, R.; FARRELL, J. A.; GENNERT, D.; SCHIER, A. F.; REGEV, A. (2015). Seurat: A practical guide. Springer.
SATIJA, R.; FARRELL, J. A.; GENNERT, D.; SCHIER, A. F.; REGEV, A. (2015). Spatial reconstruction of single-cell gene expression data. Nature Biotechnology, 33(5), 495-502.
STUART, T.A; BUTLER, Andrew; HOFFMAN, Paul; et al. Comprehensive Integration of Single-Cell Data. Cell, v. 177, n. 7, p. 1888-1902.e21, 2019. Disponível em: <https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6687398/>. Acesso em: 13 jan. 2024.
Single Cell (10X Genomics) – Centre for PanorOmic Sciences (CPOS). Cpos.hku.hk. Disponível em: <https://cpos.hku.hk/portfolio-item/single-cell-10x-genomics/>. Acesso em: 8 jan. 2024.
THEMEFISHER. SCENIC. Disponível em: https://scenic.aertslab.org/. Acesso em: 11 jan. 2024.
